Hemophilia Gene Therapy wins Conditional Approval
Part 1
Paul Clement
It has been almost 30 years since the New York Times in 1994 ran the headline “Cure for Hemophilia Is Seen by Year 2000,” regarding a prediction by the World Health Organization. But the first approval of a gene therapy for hemophilia has finally arrived!
In June, the European Medicines Agency (EMA), the European equivalent of the US Food and Drug Administration (FDA), recommended granting a conditional approval to BioMarin’s ROCTAVIANTM, the brand name of its gene therapy product, valoctocogene roxaparvovec, to treat patients with severe hemophilia A.1 On August 24, 2022, the European Commission (the EC is the executive arm of the European Union or EU) granted conditional marketing authorization to Roctavian. The EC also endorsed EMA’s recommendation for Roctavian to maintain orphan drug designation, thereby granting a 10-year period of market exclusivity, and the EC is further requiring patients treated with Roctavian to be monitored for 15 years, to ensure the long-term efficacy and safety of this gene therapy.
How does it work?
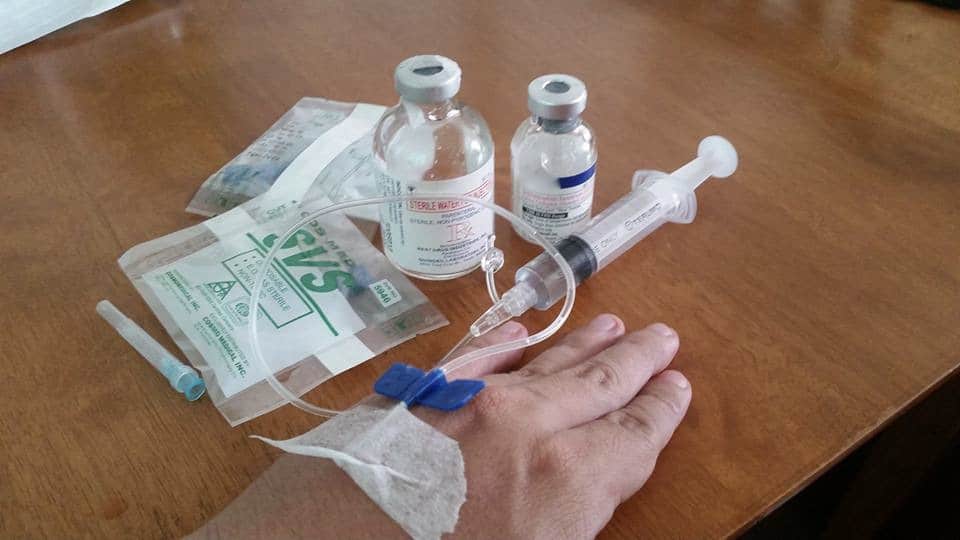
Roctavian is a single injection of trillions of copies of an adeno-associated virus (AAV) which have had their genetic material removed and replaced by a good copy of the gene for factor VIII. Once the AAV has had its genetic material removed, it can no longer replicate itself and it does not cause disease—it is now merely a transport vehicle called a “vector,” designed to get good copies of the factor VIII gene into liver cells where the liver can then produce functional factor VIII. There a many different varieties of AAV, which are given numbers to distinguish one variety from another. Roctavian uses AAV5 as a vector, which has a relatively low (4% to 50%) incidence of natural immunity (antibodies) against the vector. Roctavian does not become part of the individuals DNA and it cannot be passed on to children.
Who is eligible?
- Only adults (18 years or older) with severe hemophilia A (factor VIII deficiency) are eligible. (In children, the liver is rapidly increasing in size as the child grows, which would dilute the effect of the gene therapy).
- Only individuals who do not have factor VIII inhibitors.
- Only individuals without detectable antibodies to the vector, AAV5. The incidence of antibodies to AAV5 can vary widely, from 4% to 50%, and varies with geographic region and increases with age. (Unless the individual is immuno-suppressed, antibodies will neutralize the gene therapy treatment, rendering it ineffective.) BioMarin is running a Phase 1/2 trial, called 270-203 (ClinicalTrials.gov #NCT03520712), evaluating a single dose of Roctavian in about 10 men with severe hemophilia A who carry pre-existing antibodies against the AAV5 viral vector.
- Only individuals who do not have liver disease. (This will likely eliminate many older people with hemophilia who were infected with hepatitis C through contaminated factor products in the 1970s and early 1980s.)
Are there risks involved?
- Roctavian can cause transient inflammation of the liver, which can trigger the immune system to destroy liver cells which have the new factor VIII gene and are producing functional factor VIII. Obviously, this is not good, as it may potentially neutralize the gene therapy treatment or reduce its efficacy. This immune response can be tamped by using a corticosteroid in combination with the Roctavian infusion. BioMarin has recently completed a clinical trial, called “GENEr8-3,” of this combination treatment (Clinicaltials.gov # NCT04323098), with results expected in early 2023.
- A person’s response to Roctavian can vary widely. For many, Roctavian will not be a “cure,” but will convert a person’s severe hemophilia to mild hemophilia (factor levels between 6% and 40%).
- In clinical trials, the efficacy of Roctavian has decreased over time. This could potentially render the treatment ineffective after several years. And because the body produces antibodies against the vector after treatment, this will likely prevent the individual from receiving another gene therapy treatment using the same vector.
The very high cost of Roctavian will present challenges to its uptake, and it will also face severe competition with Hemlibra, a bispecific antibody that mimics the function of factor VIII and can be injected under the skin as infrequently as once a month. However, according to a report by the newsletter Fierce Pharma, recent market research showed that healthcare professionals in EU estimated Roctavian could capture 35% of eligible patients, in line with BioMarin’s own projection. Plus, about 80% of surveyed doctors in both EU and the U.S. expect to treat at least one patient with Roctavian within 12 months of an approval.2
In the second of this two-part series, we will look at the timeline for approval of Roctavian in the US and what it might cost.
- BioMarin is a California-based biopharmaceutical company, known as BioMarin Pharmaceutical Inc. in the US and BioMarin International Limited in Europe.
- https://www.fiercepharma.com/pharma/biomarins-hemophilia-gene-therapy-roctavian-wins-conditional-eu-backing-fda-plan-delayed