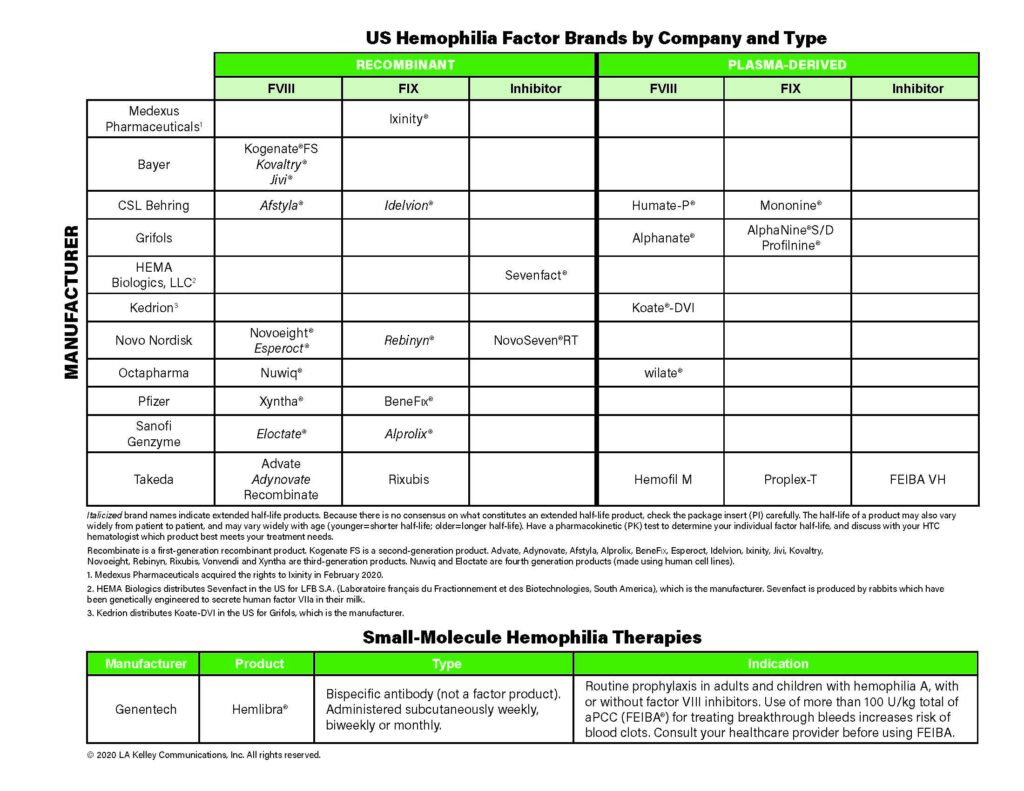
Wheels for the World 2020: A Win-Win
The pandemic has put a halt to in-person fundraisers for nonprofits like Save One Life, and yet we are extremely dependent on these events for overhead revenue. We receive program funding from grants, but for regular overhead, like salaries, rent, utilities and services, we often have to raise our own funds. Save One Life is starting to be known for its “adventure” fundraising, like cycling, mountain climbing and even hiking!

This past weekend we honored Barry Haarde, a man with hemophilia and HIV who cycled across the US six times in as many years, raising over $250,000 for Save One Life. Barry passed away in February 2018, a huge loss personally and professionally to us. He was one of a kind, a deep, sensitive soul with a heart as big as the state he lived in, Texas. We decided to first do a bike ride in 2018 to remember him, and now we continue that tradition each year to honor him and his favorite cause, helping children with hemophilia in developing countries. Save One Life was one of his top favorite charities.
Our “Wheels for the World” went virtual this year, and we opted for an in-person cycling ride (with proper masks and social distancing) starting from historic Ipswich, Massachusetts. We had eleven riders participate on Saturday to do 23 miles, including an 11-year-old! Pretty gutsy!

With lots of water and donuts, we headed out into the crisp, New England air. The course took us through the lush green farmlands and horse stables that populate this area, and past solid, magnificent colonial-style homes, some dating back to the 1600s! Along the route a small grey vole darted madly across the road, just missing my tires; a white-tail deer stood like a statue in a golden field, ears alert like radar shields as it watched us pass; and sadly, I saw a very flat chipmunk, a victim of the waning sun and probably extra body fat as he readied for hibernation.
We all returned together, and after congratulations (no hugs!), we went to True North Ale House for a complimentary beer, or in our case, blackberry Izzy drinks. I especially enjoyed chatting with Oliver, a tall young man who just started working at uniQure three weeks ago, and who is responsible for making the viral vectors into which the human gene for factor VIII is placed. He is working on the cure for hemophilia and we surely hope he is successful!
On Sunday, we reconvened on another bright, warm day, with a smaller group. I was also supposed to ride this one, 62 miles, but had woken up Friday with a back spasm. I worked it out, and was able to ride the 23 miles (which was actually 27 for us, as we got a little lost) but today, it came back with a vengeance. No way could I do 62 miles when it felt like there was a little ball with spikes sitting in my lower right back. I showed up in my gear, ever hopeful, but bailed at the last minute, my back sending warnings. I watched the six riders shove off. Jodi, Karen and I were waiting for maybe two more, and after 20 minutes a car pulled up. Was it Dan Leonard of uniQure, or Scott?

I watched as a handsome man got out of the car and approached. He said, “You don’t recognize your own brother?” My brother! All the way from the Springfield area, two hours away. I hadn’t seen him in over a year and hadn’t known he had registered! Imagine if I had not had the back spasm, and took off on the 62-mile ride. I might never have seen him that day. Jim and I hugged and chatted, and then Dan Leonard arrived. So now they both had someone to ride with. They opted for the 23 mile one that we did yesterday. And off they went!
The day was successful and I know we raised a lot of money from the wonderful sponsors, and from the dedicated riders. I didn’t even know some of the riders—two from Worcester, Massachusetts—who were college student and friends of someone in our community. They loved the ride and pledged to participate next year.
In fact, everyone had a great time. I heard from quite a few people that this year had been so strange, that they had not had a chance to ride outside at all. Injuries, work, childcare issues, COVID… this fundraising ride gave them the boost to dust off the bike (my brother’s literally had cobwebs on it!) and get out in the glorious autumn sunshine to enjoy the beauty of this state. We made new friends, reconnected with old friends, and helped Save One Life.

As Doug and I drove into downtown Ipswich, to get a bite to eat on Saturday, a tall, lean cyclist pulled right in front of us, not even staying to the side of the road, but directly in front (legally fine). I was struck by his position and body type. Incredibly, this rider had Barry’s long torso, and thin, powerful legs. The way he held his handlebars and leaned over… I had ridden enough with Barry to know his stance anywhere. It was as if he materialized to ride with us once more time…. and then he was gone. But I managed to snap a photo.
His spirit is always with us, challenging us to be better versions of ourselves, and leading us into a future where every child with hemophilia will have access to medicine. That’s why we ride, and that’s what we work for at Save One Life. RIP Barry. And thanks to everyone who sponsored, rode and supported Wheels for the World 2020!